Highlight:
UniMoMo is the first all-atom geometric latent diffusion framework capable of designing peptides, antibodies, and small molecules within a unified generative model, demonstrating superior performance and cross-domain knowledge transfer in de novo binder design.
Overview
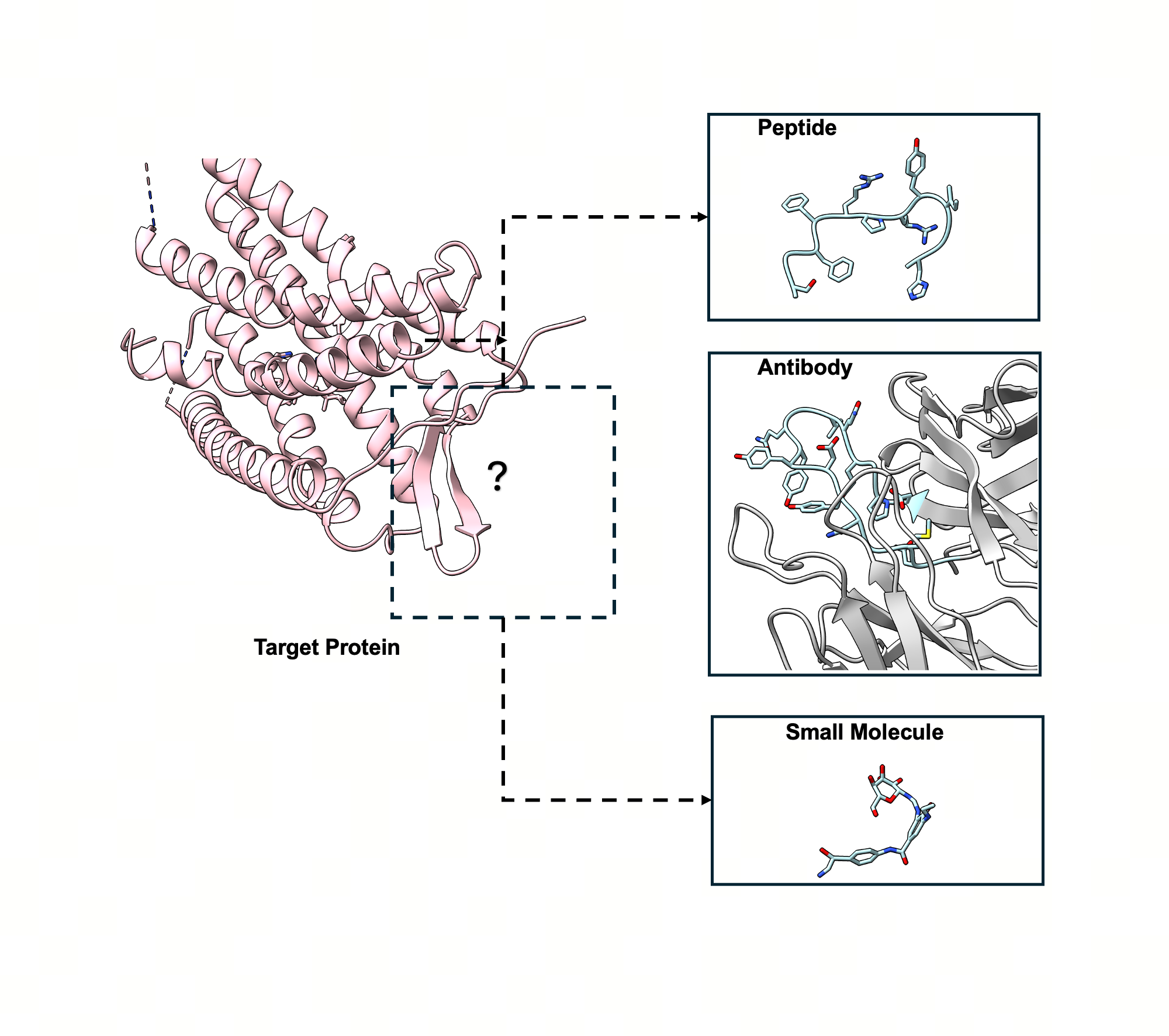
The design of target-specific binders—ranging from small molecules to peptides and antibodies—is fundamental to modern drug discovery. Historically, computational approaches have treated these molecular types as distinct domains, utilizing specialized generative models for each. This fragmentation fails to leverage cross-domain transferability and limits the exploration of versatile therapeutic strategies. We introduce UniMoMo (Unified generative Modeling of 3D Molecules), a novel framework that unifies the design of diverse molecular binders into a single, cohesive model.
Methodology
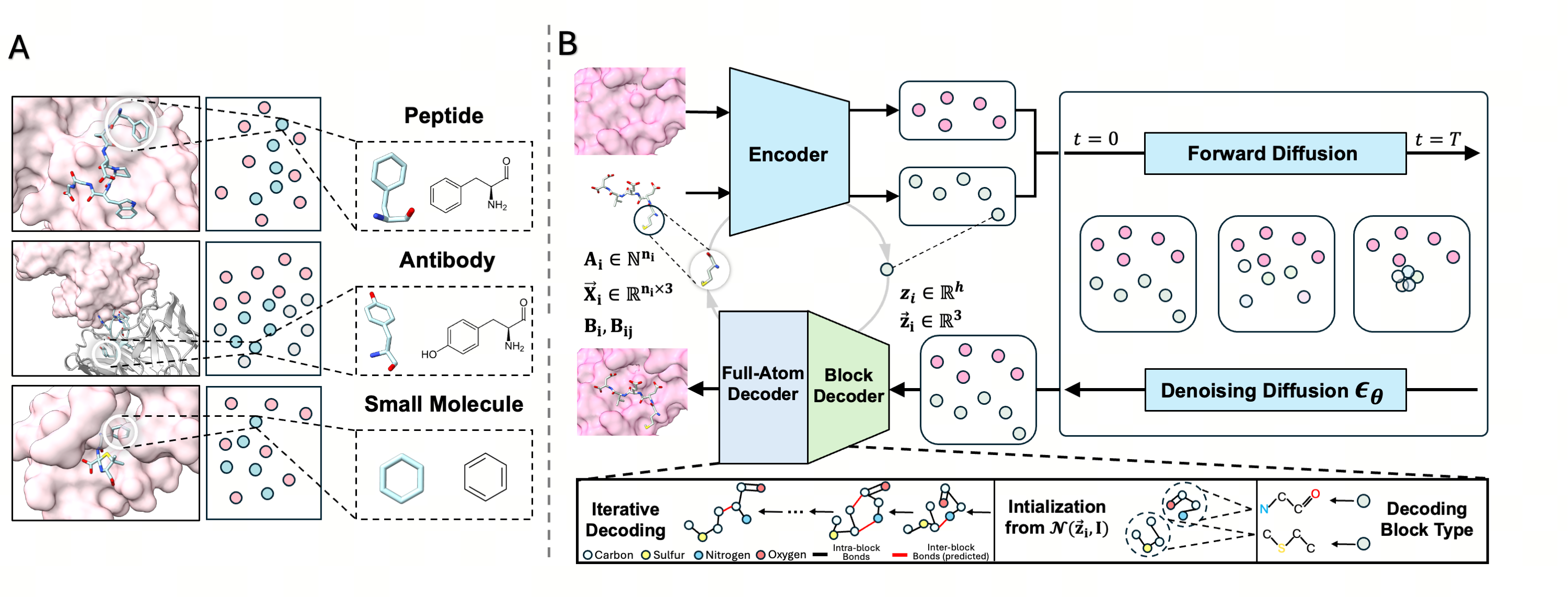
The core innovation of UniMoMo is its ability to process different biomolecular using a unified, full-atom representation called “graphs of blocks.” In this system, a block corresponds to either a standard amino acid (for peptides and antibodies) or a molecular fragment (for small molecules) identified via a principal subgraph algorithm. This representation preserves critical full-atom geometric details while capturing hierarchical structural priors. To generate new binders, the framework employs a geometric latent diffusion model. The process begins with an iterative full-atom variational autoencoder (VAE), which compresses the “blocks” into a latent space defined by low-dimensional hidden states and spatial coordinates. An E(3)-equivariant diffusion model then operates within this compressed latent space to generate the binder’s structure. This latent-space approach allows the model to focus on global structural arrangements efficiently, while a specialized decoder reconstructs the fine-grained atomic details and chemical bonds.
Performance and Cross-Domain Learning
We benchmarked UniMoMo extensively against state-of-the-art, domain-specific models across peptide, antibody, and small molecule tasks. The unified framework consistently demonstrated superior performance in key metrics, including binding energy and structural validity UniMoMo leverages the inherent advantages of multi-domain training within a unified architecture, utilizing cross-domain knowledge transfer to refine all-atom interaction patterns and geometric precision across diverse molecular classes. This approach outperformed variants trained on single domains, indicating that UniMoMo successfully learns and transfers fundamental physical principles governing molecular interactions across different chemical modalities. Furthermore, the unified training paradigm allows the model to learn a broader spectrum of property-structure relationships, mitigating the biases often seen in single-domain models . By integrating data from all molecular types, UniMoMo generates binders with highly realistic geometries, including accurate bond lengths and angles, without requiring additional post-generation relaxation by physical forcefields. This cross-pollination of data allows the model to generalize better and avoid overfitting to specific chemical spaces, resulting in molecules with superior overall performance.
Applications
The versatility of UniMoMo is demonstrated through its ability to generate high-affinity binders across multiple modalities for a single target. In a proof-of-concept study targeting a G protein-coupled receptor (GPCR), the model successfully designed peptides, antibodies, and small molecules for the same binding site . These generated candidates exhibited strong in silico binding affinities, as assessed by Rosetta and Vina scores. Notably, the unified nature of the model enabled the transfer of interaction patterns between molecular types. For instance, generated small molecules were observed mimicking the side chains of standard amino acids, such as Arginine, to form hydrogen bonds with the target protein. Additionally, the model effectively utilized local geometric motifs common in peptides and antibodies, such as amide connections, to construct wide-spanning scaffolds for small molecules . These findings highlight UniMoMo as a powerful tool for holistic drug discovery, capable of leveraging cross-domain knowledge to explore diverse therapeutic modalities for a single protein target.
Performance and Cross-Domain Learning
We benchmarked UniMoMo extensively against state-of-the-art, domain-specific models across peptide, antibody, and small molecule tasks. The unified framework consistently demonstrated superior performance in key metrics, including binding energy and structural validity UniMoMo leverages the inherent advantages of multi-domain training within a unified architecture, utilizing cross-domain knowledge transfer to refine all-atom interaction patterns and geometric precision across diverse molecular classes. This approach outperformed variants trained on single domains, indicating that UniMoMo successfully learns and transfers fundamental physical principles governing molecular interactions across different chemical modalities. Furthermore, the unified training paradigm allows the model to learn a broader spectrum of property-structure relationships, mitigating the biases often seen in single-domain models . By integrating data from all molecular types, UniMoMo generates binders with highly realistic geometries, including accurate bond lengths and angles, without requiring additional post-generation relaxation by physical forcefields. This cross-pollination of data allows the model to generalize better and avoid overfitting to specific chemical spaces, resulting in molecules with superior overall performance.
Applications
The versatility of UniMoMo is demonstrated through its ability to generate high-affinity binders across multiple modalities for a single target. In a proof-of-concept study targeting a G protein-coupled receptor (GPCR), the model successfully designed peptides, antibodies, and small molecules for the same binding site . These generated candidates exhibited strong in silico binding affinities, as assessed by Rosetta and Vina scores. Notably, the unified nature of the model enabled the transfer of interaction patterns between molecular types. For instance, generated small molecules were observed mimicking the side chains of standard amino acids, such as Arginine, to form hydrogen bonds with the target protein. Additionally, the model effectively utilized local geometric motifs common in peptides and antibodies, such as amide connections, to construct wide-spanning scaffolds for small molecules . These findings highlight UniMoMo as a powerful tool for holistic drug discovery, capable of leveraging cross-domain knowledge to explore diverse therapeutic modalities for a single protein target.