Highlight:
This study presents ImmunoSeq, an interpretable and applicable method for immunogenicity prediction. ImmunoSeq demonstrated superior ADA correlation and humanness classification accuracy compared to deep learning models, while accurately predicts ADA reductions in humanization, enabling sufficient sequence optimization for humanness.
Overview
Immunogenicity remains a critical bottleneck in biotherapeutic development. A central challenge in immunogenicity prediction lies in identifying T cell epitopes presented by major histocompatibility complex (MHC) molecules. However, the MHC system is highly polymorphic, and T cell receptor (TCR) recognition is highly context-dependent, making it difficult to accurately predict which peptides will trigger an immune response. Current immunogenicity prediction methods fall into distinct categories, each with notable limitations. We present ImmunoSeq, rooted in the biological principle of immune tolerance: the human immune system is tolerant to self-proteins, which do not elicit ADA.
Methodology
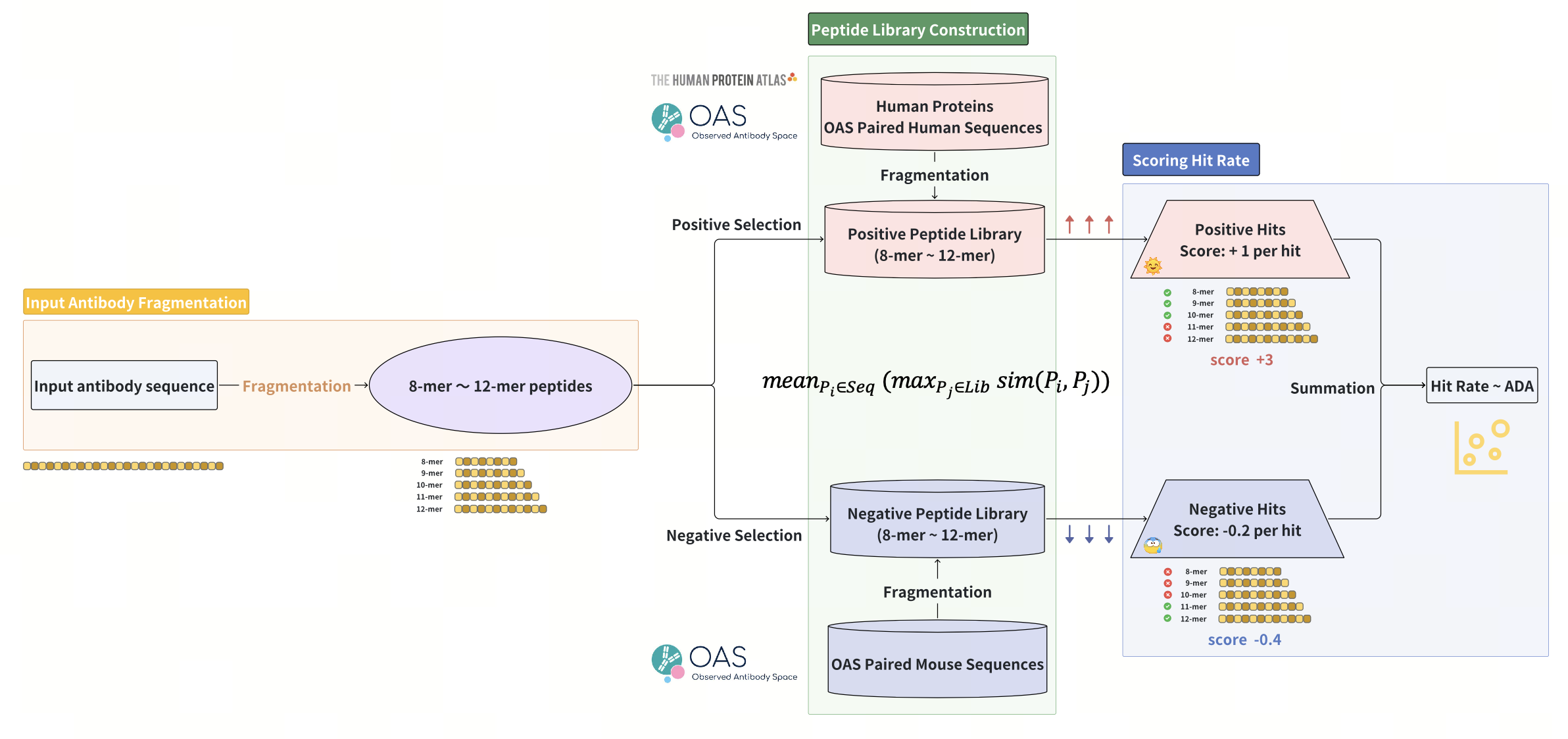
ImmunoSeq constructs a self-peptide library by systematically fragmenting >20,000 human protein sequences and >1 million healthy human antibody sequences from the OAS paired dataset into k-mer peptides (k=8-12, matching dominant MHC-presented lengths), thereby generating a virtual repository of millions of immunologically safe peptides. Complemented by a negative non-self-peptide library derived from >80,000 mouse antibody sequences also from OAS paired dataset, this approach incorporates evolutionary selected immunogenic peptides for penalty calibration. For candidate therapeutic antibodies, we similarly fragment their sequences into k-mer peptides, quantifying both positive hits (matches against the self-peptide library) and negative hits (matches against the non-self-peptide library), assigning + 1.0 to each positive hit and -0.2 to each negative hit. These accumulated values of all hits, normalized against the total peptide number, yield the global hit rate. Theoretically, a higher hit rate indicates greater similarity to self-proteins, thus predicting lower ADA risk. Critically, ImmunoSeq achieves residue-level resolution of immunogenic contributions by quantifying per-residue hit rate – calculated as the sum of assigned values across all k-mer peptides containing the residue, normalized against the total peptide number. Residues with low hit rate are flagged as potential immunogenic hotspots, enabling precise localization of risky regions and guiding iterative mutation design to enhance the overall hit rate.
Performance
ImmunoSeq correlates well with ADA incidence and accurately predicts immunogenicity changes during humanization.
ImmunoSeq exhibited comparable accuracy to DL methods in classifying human sequences across different species.
ImmunoSeq shows superior practical utility in iterative sequence optimization: when generating mutation candidates to boost overall hit rate, model-recommended mutation sets aligned closely with experimental observations in 25 humanized antibody pairs. Specially, the top-ranked mutations (e.g., top 5 of recommendations) were highly concordant with experimentally validated beneficial mutations.
Resources The ImmunoSeq code is available at GitHub