Highlight:
This study establishes a comprehensive benchmarking framework for in silico gene perturbation, systematically evaluating ten AI methods across four biological scenarios to standardize assessment and advance computational drug discovery.
Overview Understanding how gene expression responds to perturbations is fundamental to decoding cellular functions and identifying therapeutic targets. While high-throughput technologies like Perturb-seq have advanced this field, experimentally testing every gene perturbation across all cell types remains practically infeasible due to cost and biological constraints. Consequently, artificial intelligence (AI) models for in silico perturbation have emerged as a solution to predict cellular responses. However, the development of these models has been hindered by the lack of a standardized evaluation framework, with methods often tested on limited datasets and inconsistent metrics.
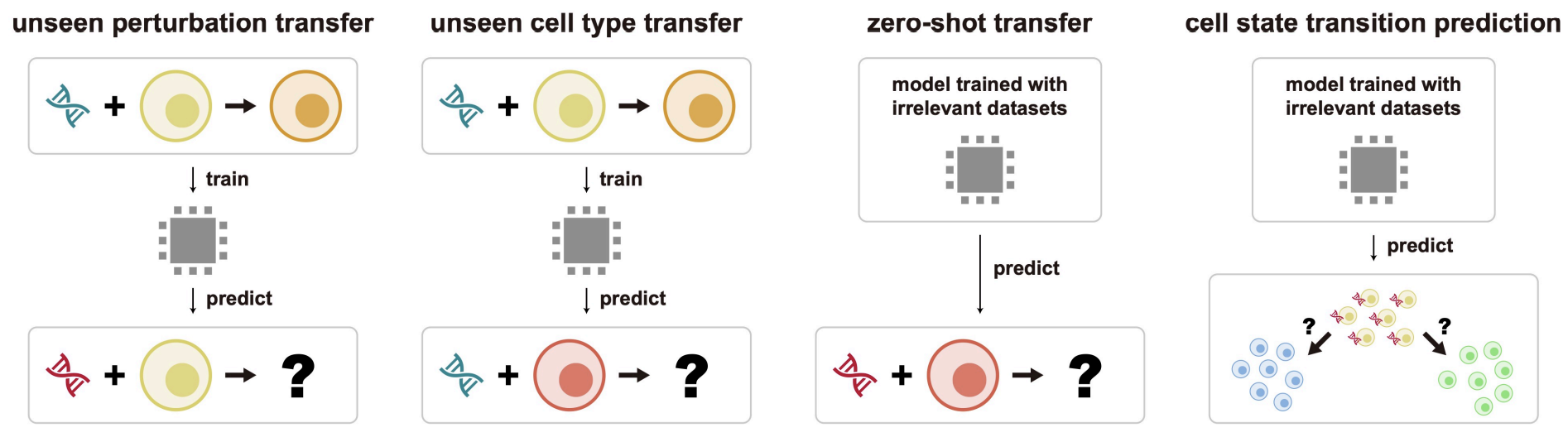
Benchmarking Framework and Scenarios To address this challenge, we introduced a systematic benchmarking framework that evaluates methods across four distinct, biologically relevant scenarios:
- Unseen Perturbation Transfer: Predicting the effects of novel perturbations within known cell types.
- Unseen Cell Type Transfer: Predicting known perturbations in previously unobserved cell types.
- Zero-Shot Transfer: Applying models to predict perturbation effects without task-specific training data, validated against bulk RNA-seq (CMAP) datasets.
- Cell State Transition Prediction: Assessing the ability to predict key gene drivers that induce transitions between specific biological states.
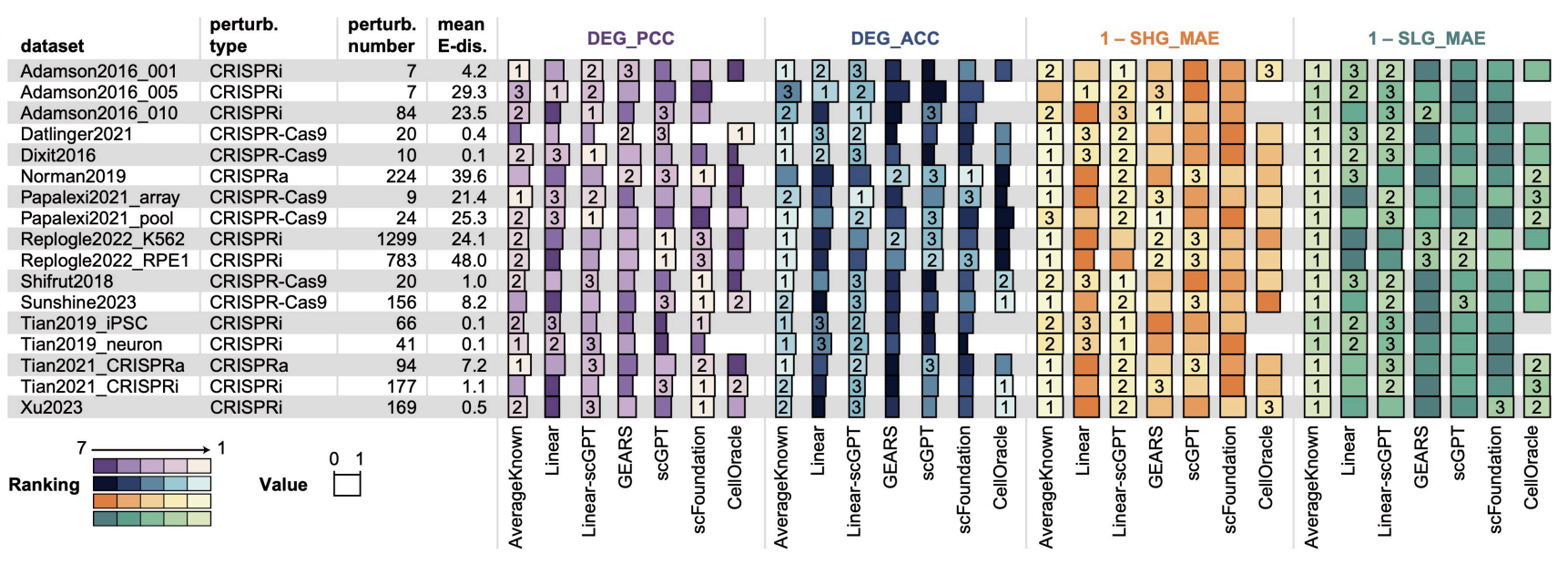
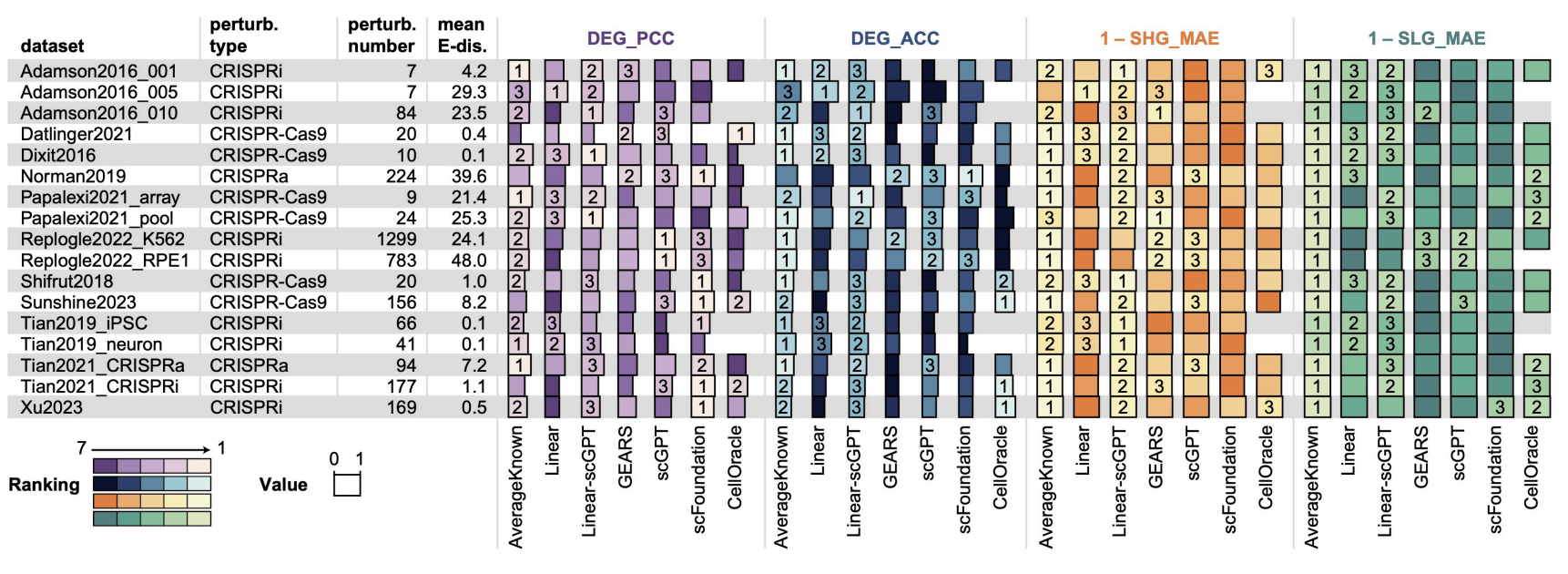
To support this framework, we curated and standardized 17 single-cell RNA sequencing (scRNA-seq) datasets, encompassing approximately 984,000 cells and 3,190 gene perturbations. We also implemented a rigorous set of metrics, including Pearson correlation coefficients and direction accuracy for differentially expressed genes, to ensure objective performance assessment.
Key Findings We benchmarked ten distinct methods, ranging from linear baselines to advanced single-cell foundation models like scGPT and scFoundation. Our analysis revealed that no single method consistently outperformed others across all scenarios:
- Foundation Models: In the “Unseen Perturbation Transfer” scenario, large pretrained models (scGPT, scFoundation) demonstrated superior performance, effectively capturing significant gene expression changes when sufficient training data was available.
- Generalization Challenges: For “Unseen Cell Type Transfer,” advanced deep learning models frequently struggled to outperform simple baseline methods (DirectTransfer), highlighting significant difficulties in generalizing across distinct cell lineages.
- Zero-Shot Limitations: The “Zero-Shot Transfer” scenario remains a substantial challenge for the field, with most current methods showing limited predictive capability compared to random baselines.
- State Transitions: For predicting cell state transitions, correlation-based methods and CellOracle proved most effective for continuous trajectories, while predicting discrete state shifts remains difficult for all tested architectures.
Impact and Resources This work provides a critical reference for the computational biology community, defining the capabilities and limitations of current AI approaches. To facilitate future research, we have released CellPB, a Python module that allows researchers to easily access these benchmarking datasets and evaluate their own models. This framework serves as a foundation for bridging computational predictions with experimental validation, ultimately supporting the development of more robust AI tools for therapeutic discovery.