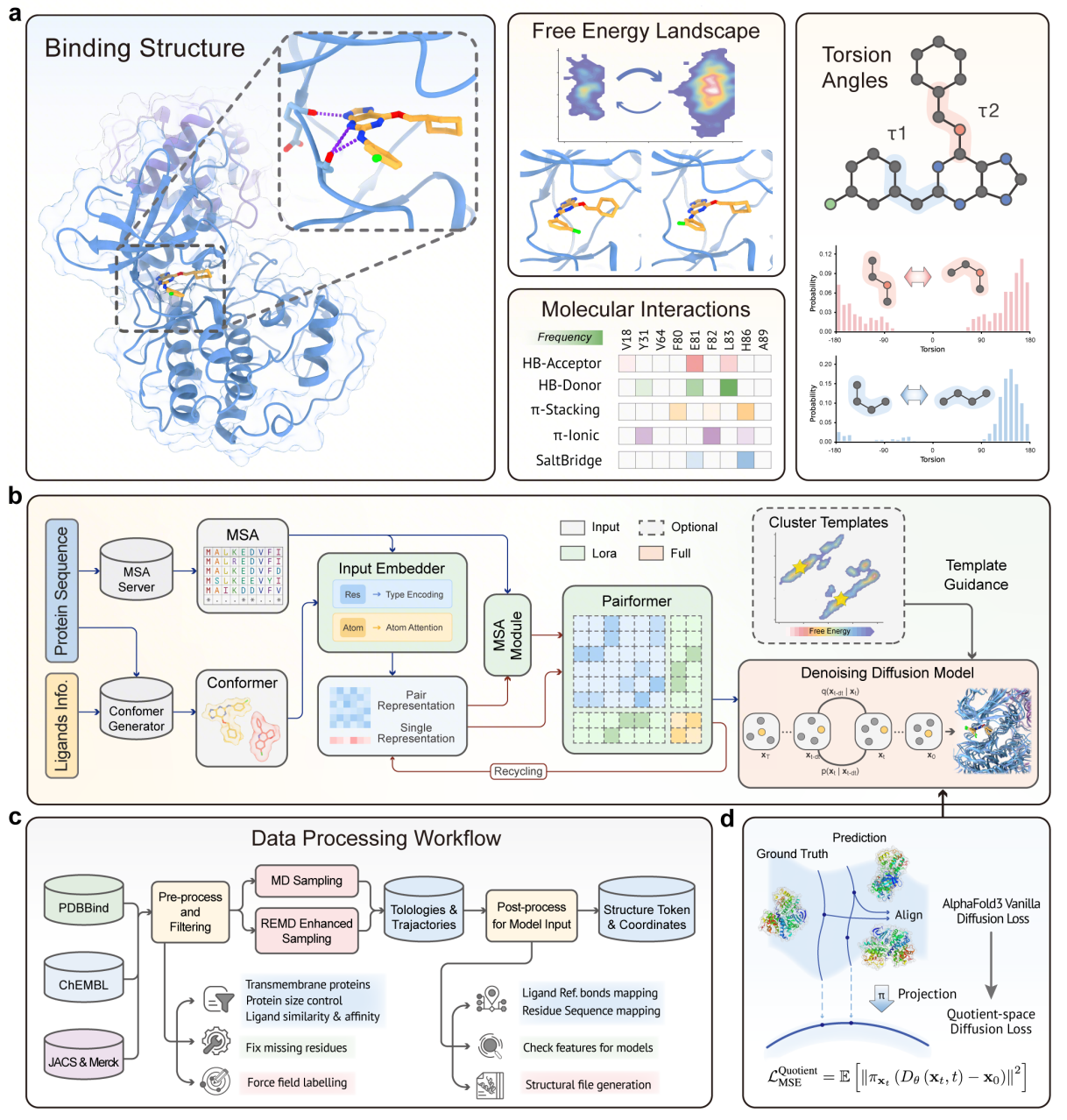
Introduction
Over the past few years, artificial intelligence has dramatically advanced protein structure prediction, pushing sequence-to-structure accuracy to unprecedented levels. However, for real drug discovery and the molecular systems it aims to understand, a single static structure is far from sufficient.
Protein–ligand binding is fundamentally a dynamic process: conformations continuously shift, energy states exchange, and critical interactions form and dissolve across multiple timescales. Understanding this dynamic thermodynamic landscape has long been a significant hurdle in drug discovery.
Today, the Anew Therapeutics research team is thrilled to introduce AnewSampling — the first systematic model designed to perform dynamic equilibrium sampling of protein–ligand complexes.
While existing co-folding models stay focus on predicting a single static structure, AnewSampling brings generative AI into all-atom thermodynamic sampling. By efficiently generating conformational ensembles that closely match molecular dynamics (MD) distributions, it pushes AI-driven drug discovery into its next phase.
The Real Molecular World Is Never Static
For many challenging targets, a single predicted structure is not enough. In practice, therapeutic behavior is rarely determined by a single binding pose alone, but the broader conformational ensemble and the equilibrium relationships within it.
If static structure prediction is like capturing a high-resolution photograph of a complex, the underlying molecular reality is closer to a continuously evolving film. AnewSampling moves AI beyond predicting isolated structures and toward modeling the dynamic conformational landscapes of protein-ligand complexes.
AlphaFold gave AI a view of molecular structure, AnewSampling gives it access to molecular motion.
From Expensive Simulation to Scalable Modeling
In structure-based drug design—particularly during lead optimization—the most valuable information often includes:
- The binding modes of ligands within the pocket
- Coupled motions between key residues and pharmacophoric groups
- Rare but functionally important low-frequency states
- Dynamic interaction networks that influence stability, selectivity, and activity
These signals are precisely what static models struggle to provide. Although classical molecular dynamics simulations can reveal them, their computational cost often limits large-scale deployment.
AnewSampling brings rich dynamic and thermodynamic signals, which are once accessible only through costly simulations, into everyday drug discovery workflows. For research teams, that means:
- Faster insight: performing equilibrium sampling at a fraction of the time required by traditional MD.
- Broader reach: generalizing across protein families and ligand chemistries without repeated retraining for each target.
- Deeper understanding: generating conformational ensembles rather than single predictions, enabling the discovery of hidden but functionally important states.
AnewSampling does more than accelerate dynamic modeling. It replaces a major step that once depended on expensive, serial physical simulation with a scalable AI system that can be deployed far more broadly. This marks an important step for the field. AI is beginning to model how complexes move, which states are thermodynamically favored, and how transitions unfold across an energy landscape.
The move from static structures to dynamic distributions marks a meaningful turning point for AI-driven drug discovery.
Superior Performance for Advanced Applications
AnewSampling delivers strong performance across a broad set of rigorous evaluations, with results that translate directly into real-world application for drug discovery.
Validated across diverse benchmarks
Across an internally constructed test set, the public JACS and Merck benchmarks, and the large-scale protein dynamics dataset ATLAS, AnewSampling demonstrates superior performance.
The conformational ensembles it generates for both protein monomers and protein–ligand complexes closely match reference MD simulations, indicating robust generalization across unseen targets, diverse ligand chemistries, and different evaluation settings.
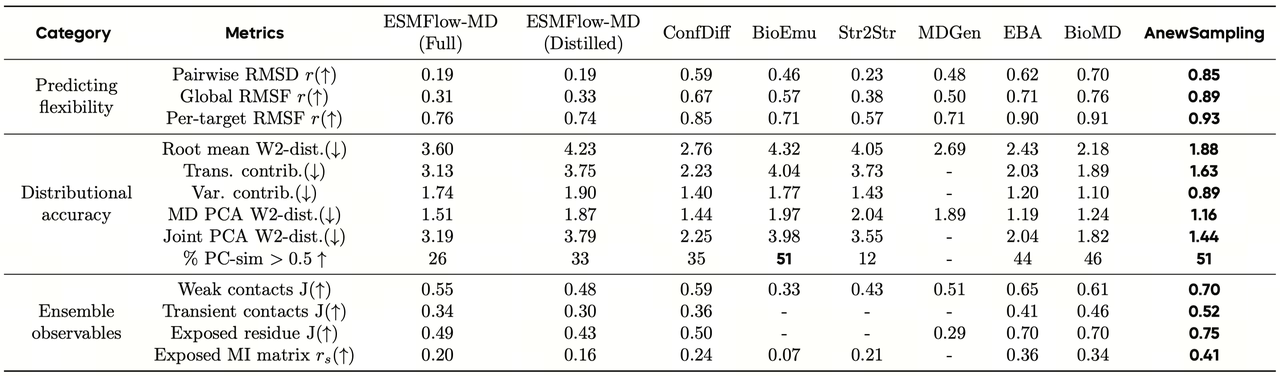
High-fidelity dynamics where they matter
More importantly, AnewSampling’s advantages extend beyond producing structures that merely “look similar.” The model recovers dynamic signals that directly influence drug discovery decisions.
Across key metrics such as ligand torsion distributions, protein–ligand interaction networks, and protein flexibility changes, AnewSampling significantly outperforms existing generative models while remaining highly consistent with MD references. This suggests the model is not simply capturing superficial geometric similarity, but rather learning distributions closer to the underlying thermodynamic reality.
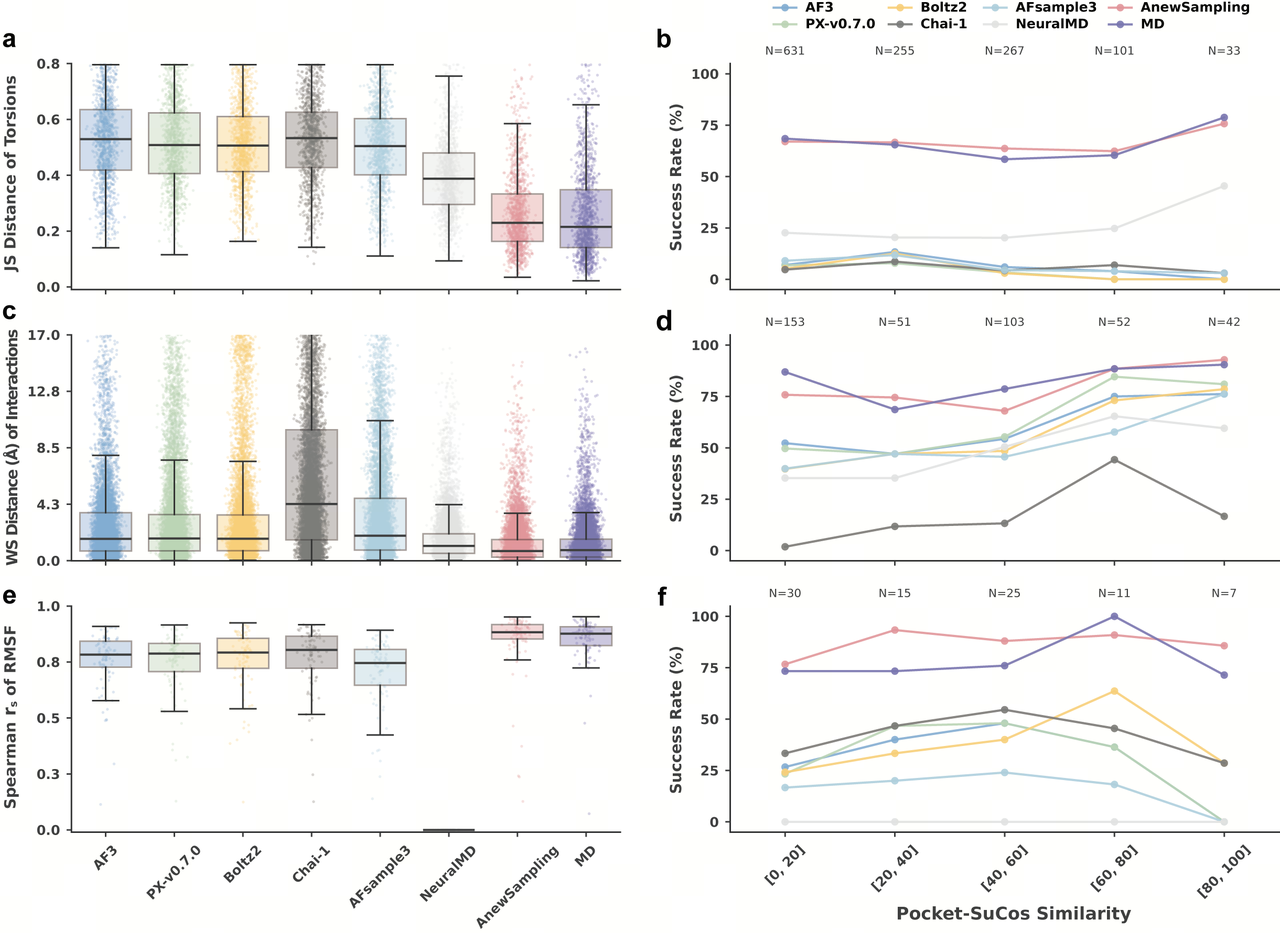
Further analysis using the JACS and Merck datasets shows that AnewSampling achieves significantly higher success rates in generating protein–ligand conformational landscapes than existing static or dynamic prediction models, while maintaining strong correlation with MD results.
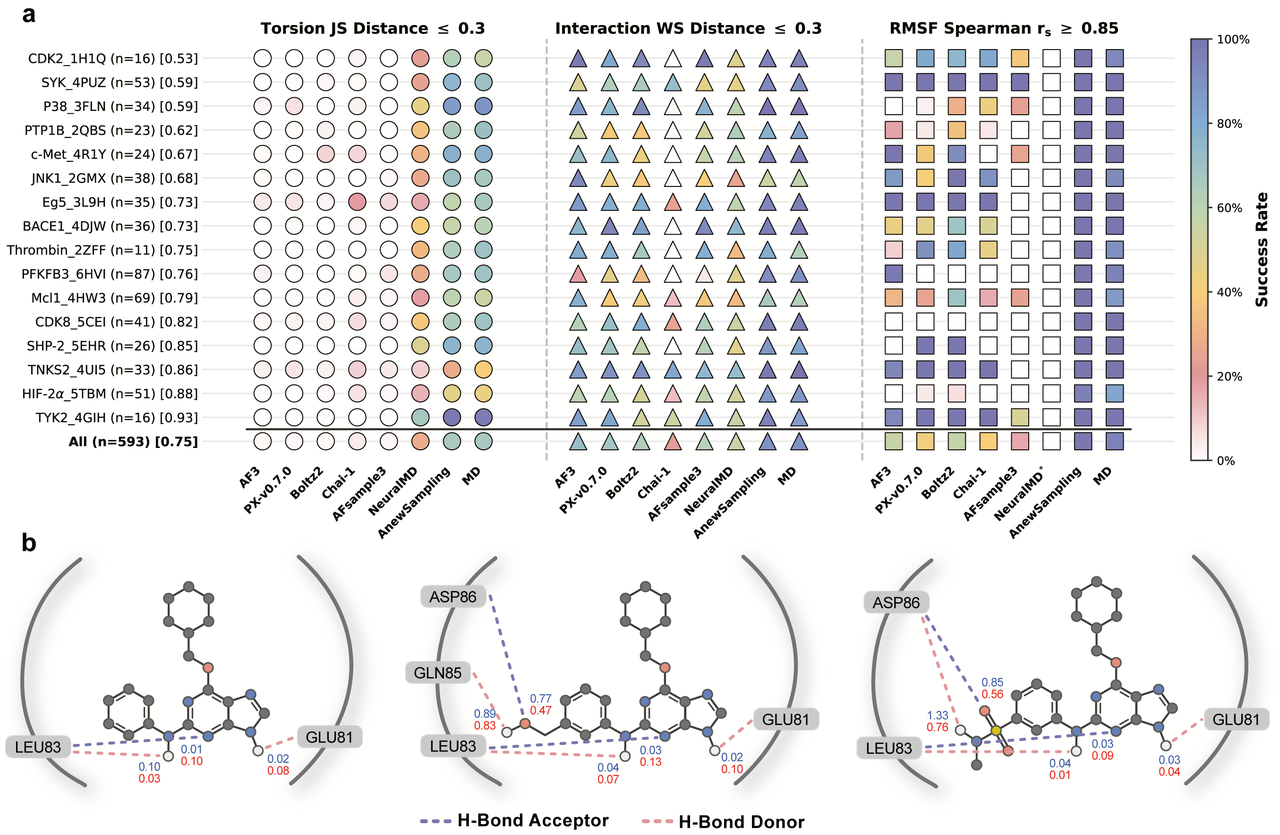
Stronger coverage of functional states:
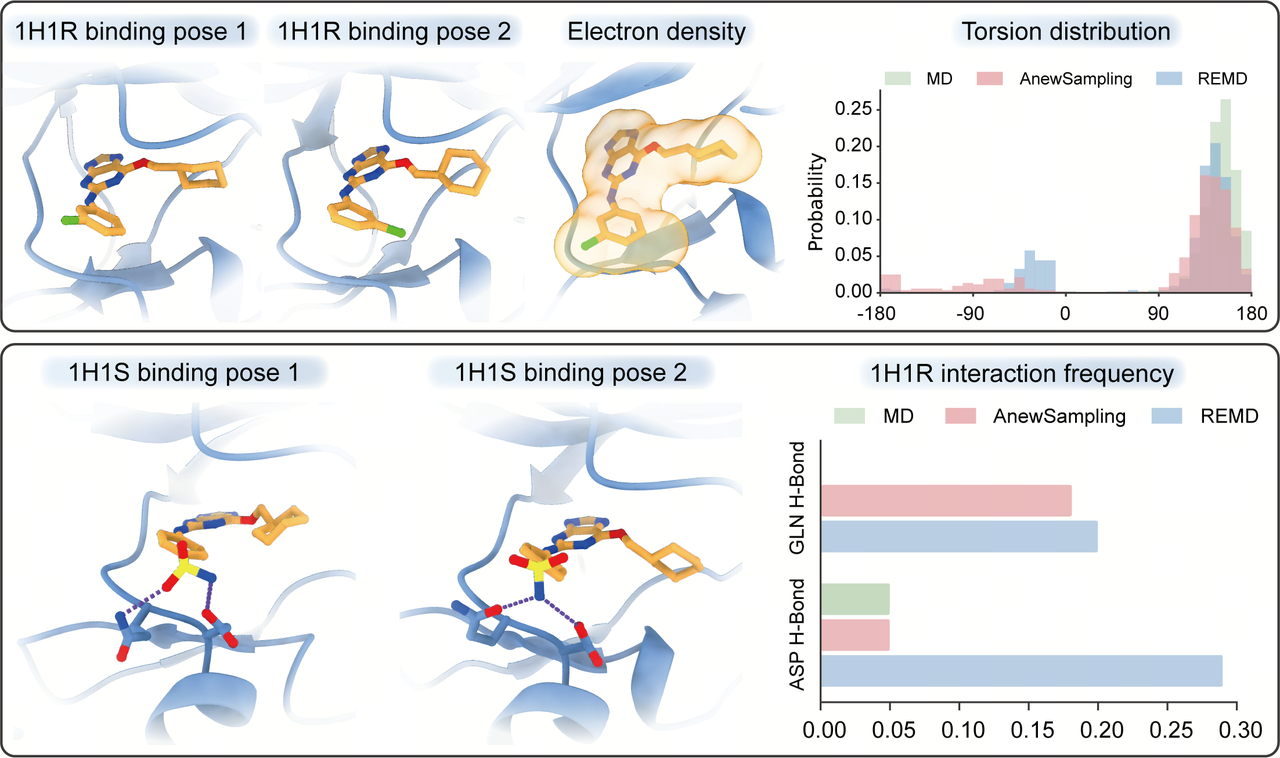
The most revealing tests are often the challenging ones. In the CDK2 system, AnewSampling captures diverse binding modes and complex cooperative motions between ligands and side chains—states that are often difficult to sample sufficiently with conventional MD under standard simulation budgets.
In this case, AnewSampling achieves performance approaching that of enhanced sampling methods such as Replica-Exchange Molecular Dynamics (REMD). This suggests that functional states previously accessible only through higher-cost simulations can now be explored efficiently with AnewSampling.
Toward a New Era of Dynamics-Aware AI Drug Design
The emergence of AnewSampling marks an important step forward—from structure prediction toward dynamic molecular understanding.
If earlier AI systems helped us see what molecule complexes look like, the next generation of models is learning how molecules move, evolve, and how those dynamics determine biological function and druggability.
By establishing a new balance between speed, scale, and physical fidelity, AnewSampling provides a powerful new foundation for exploring protein–ligand interaction landscapes and expands the possibilities for future drug discovery applications.
Molecular dynamics information that once required costly simulations is now becoming increasingly accessible at scale.