Redefine Feasibility in Medicine
Making Previously Unreachable Therapies Achievable.
AI matters most where conventional drug discovery falls short.
That is where Anew Labs is built to focus
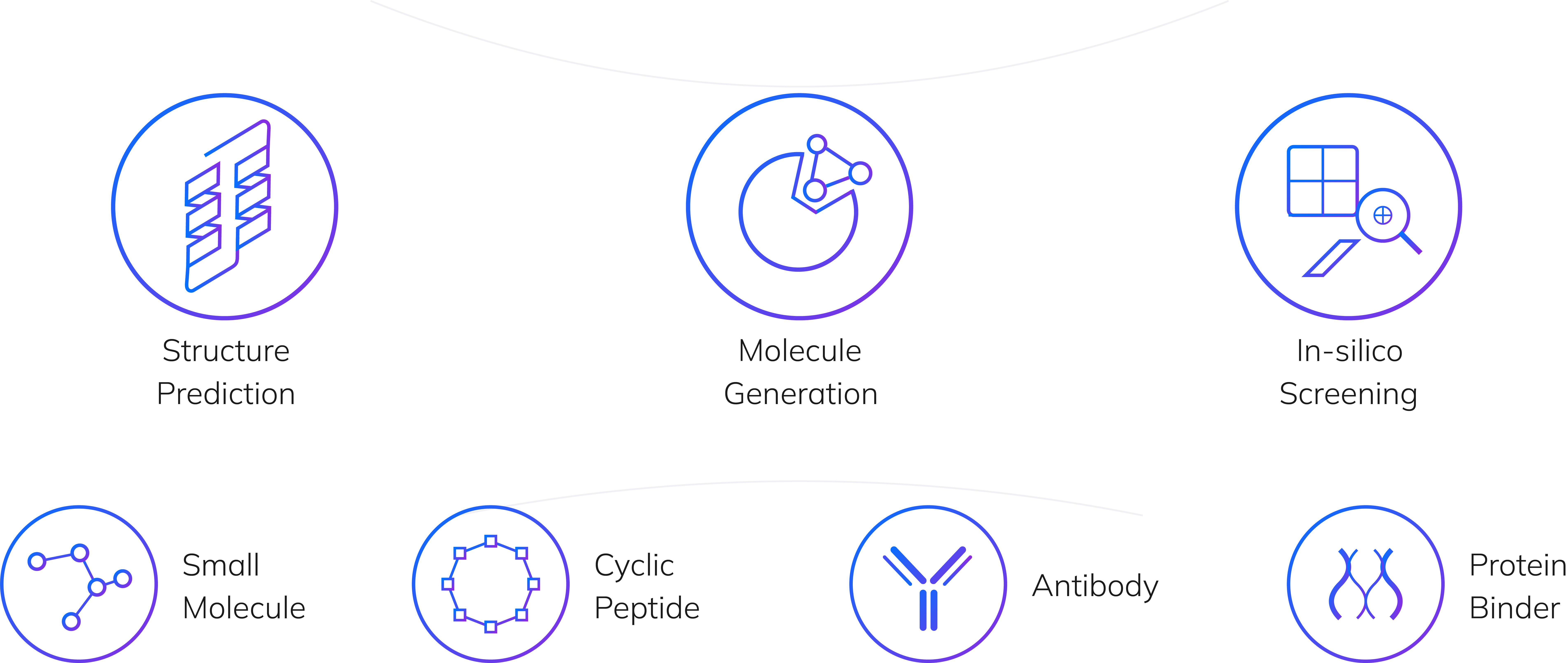

Research
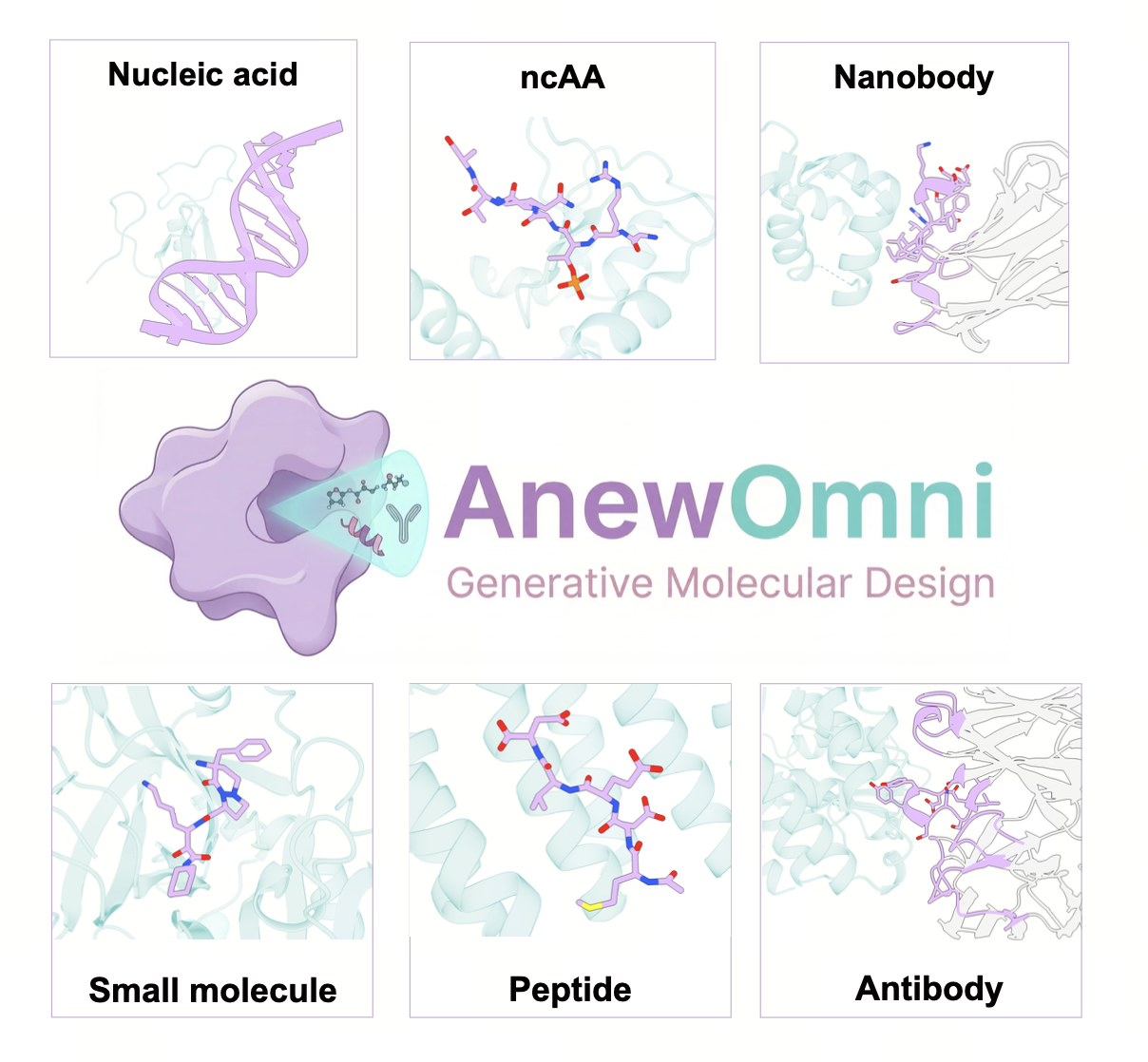
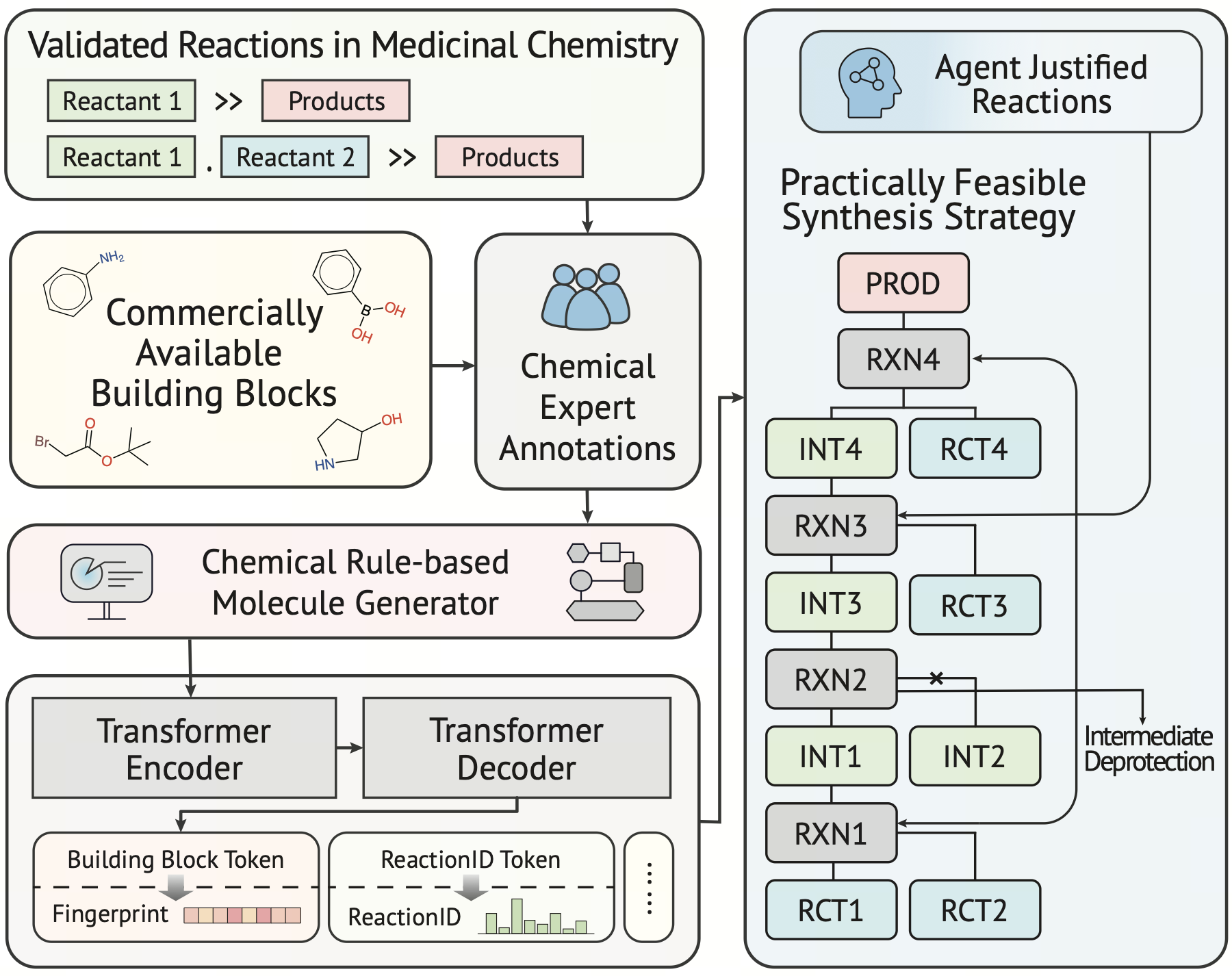
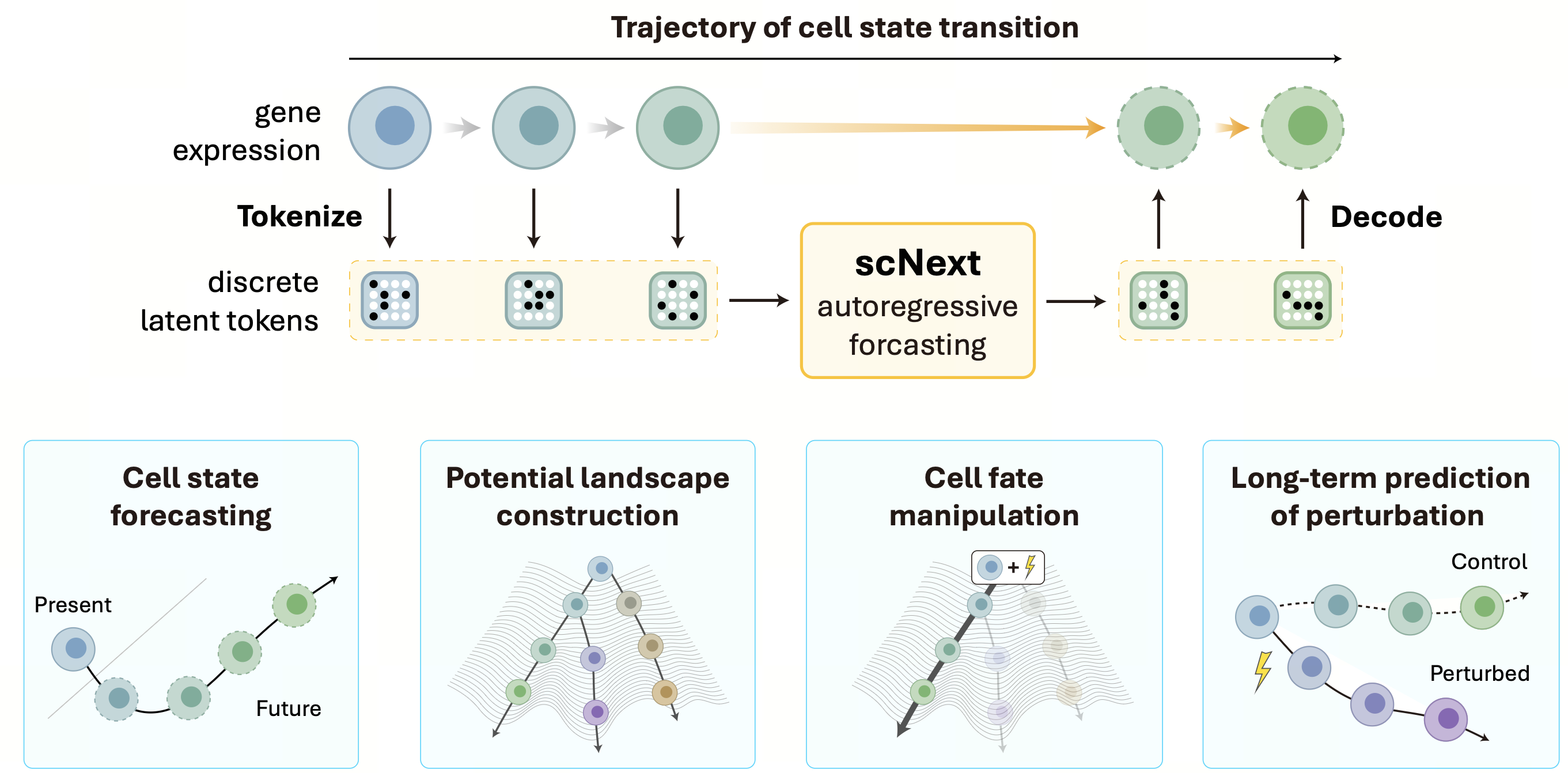
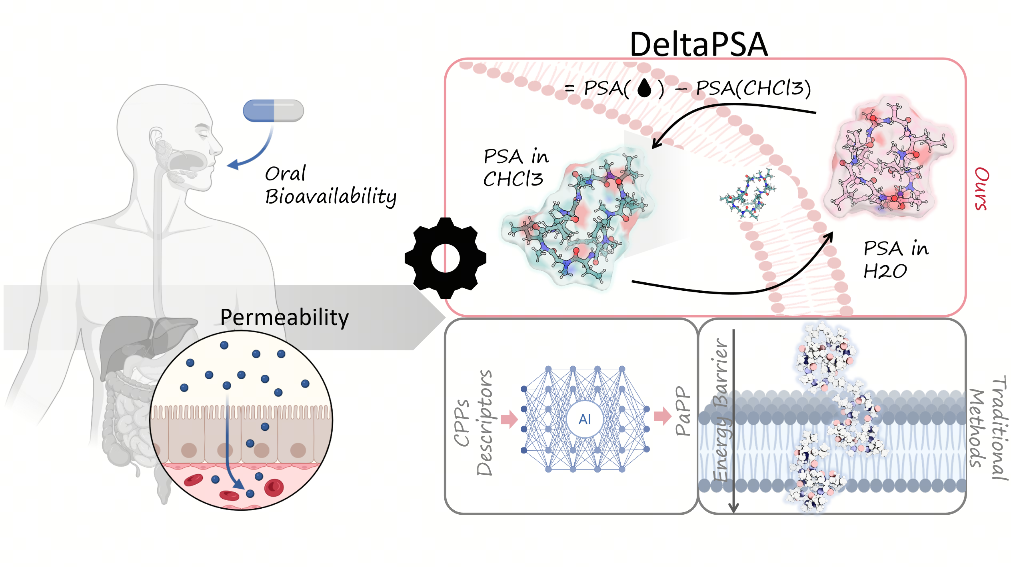
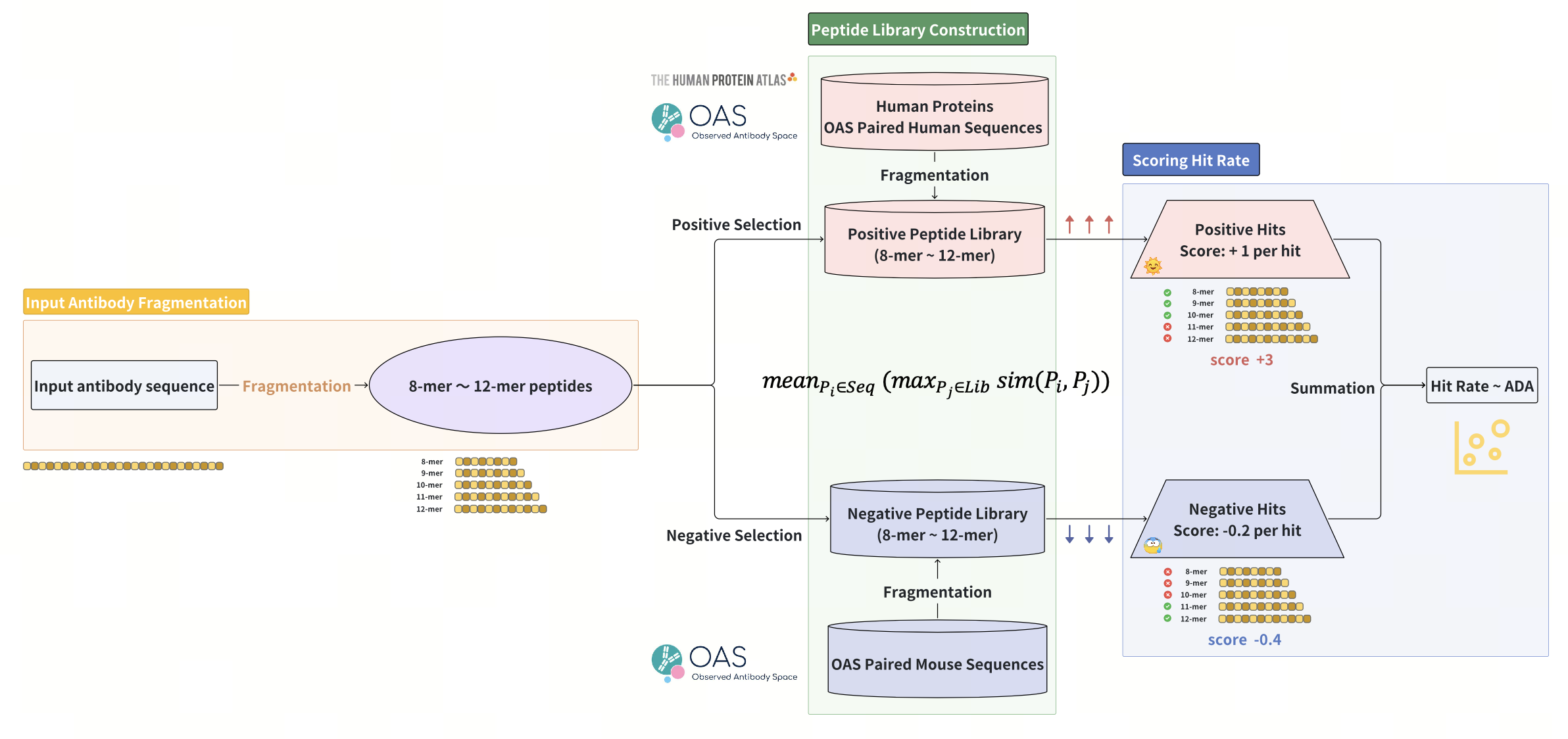
Pipelines
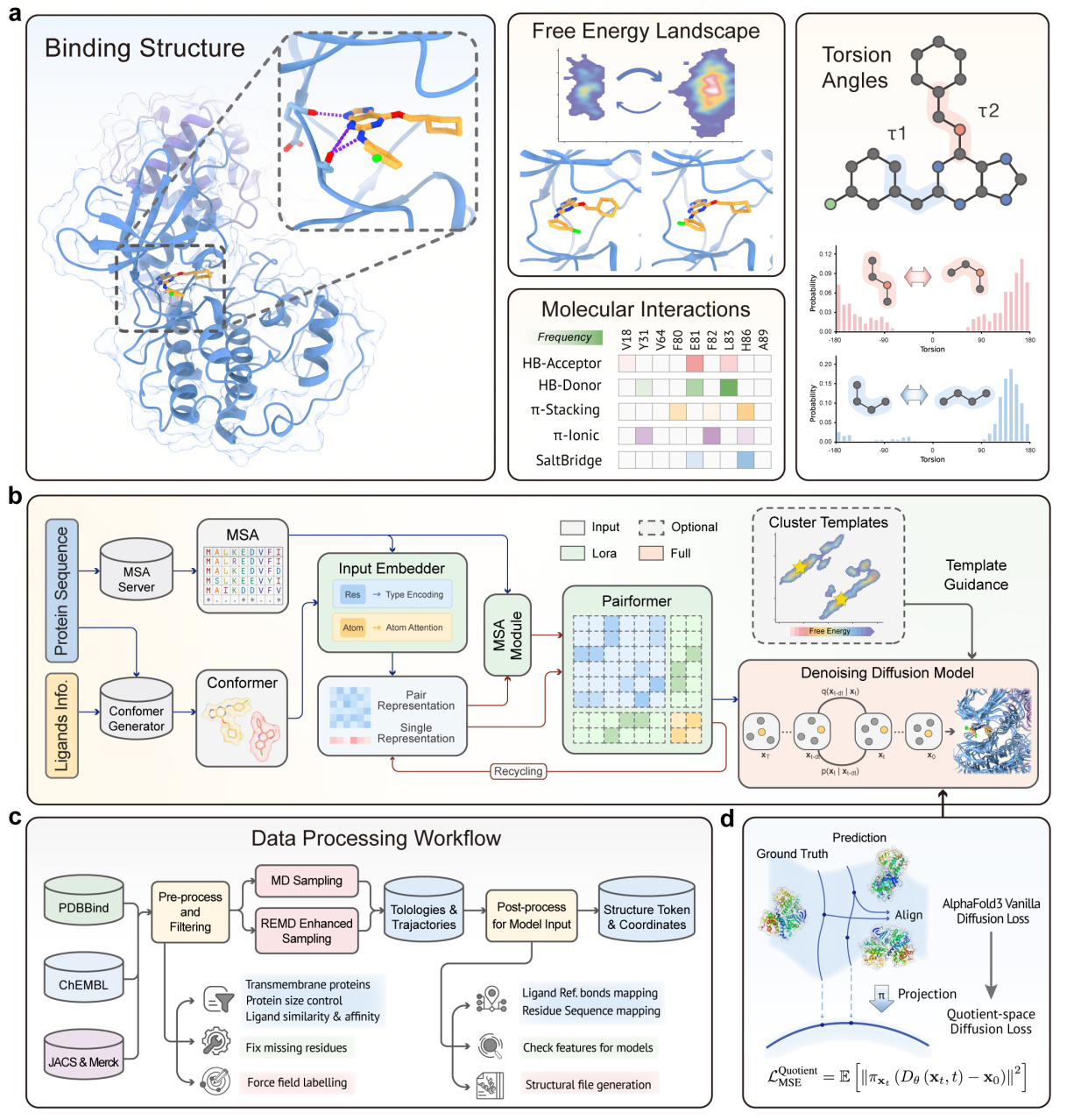
Our integrated AI platform accelerates every stage of drug discovery, from target identification to clinical development.
Exploratory
Hit ID
Hit to Lead
Lead Optimization
IND Enabling
IL17AA/AF/FF
IL4R
Undisclosed Target 1
Undisclosed Target 2
Completed
In Progress
Planned
Team
Core Team* By joining date

Kai Liu

Qiaojing Huang

Weiyang Dai

Haoyu Yu

Yuli She

Wentao Li

William Hilbert

Jiahui Tong

Haoxiang Gao

Chris Li

Yan Shi

Zhiye Guo

Gaodeng Li

Zhihong Zhou

Junwei Chen

Cheah Chen Seh

Xuan Wu

Meng Wu

Shi Jie Teo

Ziting Zhang

Qilong Wu

Huidong Yu

Nanjun Chen

Youjun Xu

Xiangzhe Kong

Yiping Yu

Qingyuan Zhu

Wenjuan Tan

Shaoning Li

Yusong Wang

Songyang Wang

Pengkang Guo

Rui Jiao

Ziyi Yang

Ran Yang
Scientific Advisory Board

Yongjun Liu

Wei Zhu

Ji Ma

Richard Miller

Hua Zou

Xiaolun Wang