Highlight:
Cost-Effective Prediction of Macrocyclic Peptide Permeability via Dynamic Conformational Analysis We introduce a computationally efficient molecular dynamics framework that accurately predicts macrocyclic peptide permeability by quantifying environment-dependent conformational adaptability and specific shifts in polar surface area .
Mechanistic Insights and Predictive Modeling of “Chameleonic” Cyclic Peptide Permeability
The clinical potential of cyclic peptides is often limited by poor membrane permeability, a property that is difficult to predict due to the complex conformational flexibility of these molecules. To address this challenge, we developed a comprehensive framework that integrates molecular dynamics (MD) simulations with machine learning to decipher the mechanistic determinants of passive membrane transport.
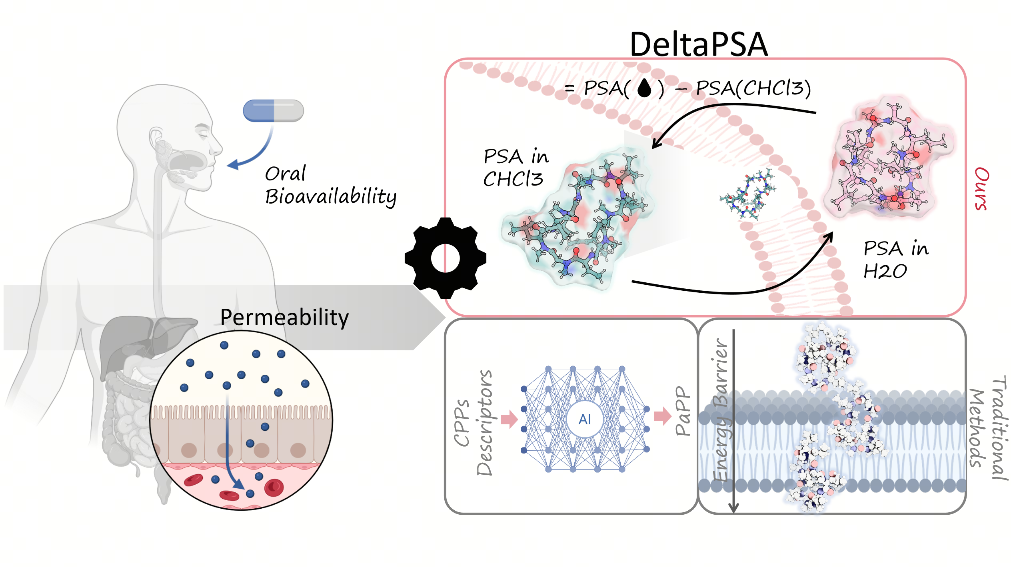
Our approach addresses the limitations of conventional static descriptors by explicitly modeling the dynamic behavior of peptides in distinct solvent environments. We utilized MD simulations in both aqueous and chloroform solvents to mimic the polarity gradient encountered during membrane translocation. By employing high-temperature simulations (490 K), we enhanced the sampling of rare conformational transitions, allowing us to capture the “chameleonic” nature of macrocyclic peptides—their ability to expose polar groups in water while shielding them in nonpolar membrane environments.
A key finding of this study is the identification of a size-dependent threshold for conformational adaptability. Our analysis reveals that macrocyclic peptides with a monomer length of nine or more residues exhibit significant solvent-dependent conformational switching. For these larger macrocycles, the difference in polar surface area (ΔPSA) between aqueous and membrane-mimicking states serves as a critical quantitative indicator of permeability. Peptides capable of minimizing ΔPSA through structural reorganization demonstrate lower energetic costs for membrane insertion and higher permeability rates.
Building on these insights, we trained machine learning models to predict permeability outcomes. A Random Forest classifier achieved an F1-score of 0.86, demonstrating high predictive accurac. Feature importance analysis identified two specific solvent-accessible surface area (SASA) metrics—nonpolar SASA in aqueous solvent and polar SASA in chloroform—as the most potent predictors of membrane permeability. These descriptors directly quantify the peptide’s capacity to balance solubility and lipophilicity through dynamic structural adjustments.
Furthermore, we optimized the computational workflow for high-throughput application. We demonstrated that two independent 300-ns simulations are sufficient to extract robust dynamic features, substantially reducing the computational expense compared to traditional enhanced sampling methods. This framework provides Anew Therapeutics with a scalable, mechanistically grounded tool for the rational design of cell-permeable macrocyclic therapeutics.